Main protease of the SARS-CoV-2 is an important drug target in the effort of finding potential inhibitors of the SARS-CoV-2 virus. In this short article, we will try to do a simple 3D visual analysis of it’s binding site based on recent published research and discovery. We provide a short summary of the SARS-CoV-2 main protease binding site; a simplified view of the protease binding site and it’s surrounding area.
The 3CL Mpro structure is composed of three domains:
Domain I: residue 8 – 101
Domain II: residues 102 – 184 , which is the catalytic domain
Domain III: residues 201 – 303 which is responsible for the enzyme dimerization [1]
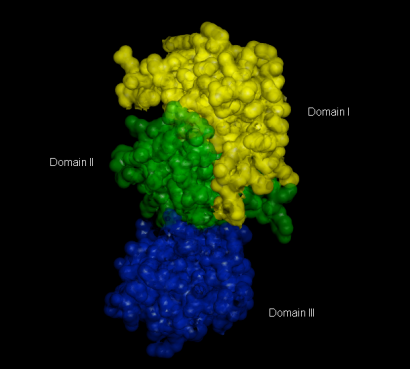 Figure 1. 3D Surface Representation of Domain I to III of the Main Protease Mpro (PDB ID: 6LU7)
Figure 1. 3D Surface Representation of Domain I to III of the Main Protease Mpro (PDB ID: 6LU7)
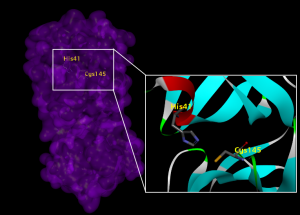
Figure 2. The catalytic dyad consist of Cys145 & His45 from Domain I & II (PDB ID: 6LU7)
Some researchers [1],[2],[3] have categorized the active site cavity into subsets S1 – S5 as follows:
S1: His163, Glu166, Cys145, Gly143, His172, Phe140
S2: Cys145, His41, Thr25
S3 – S5: Met49, His41, Met165, Glu166, Gln189
The subsite S1′ is located on the opposite site of S1 [5],[6] where the cleavage site is between the P1 and P1′ group [6]. Subsite S1, S2, and S4 are shaped into well defined binding pockets, whereas S3, and S5 are located on the protein surface which is more accessible to solvents. [3]

Figure 3. Surface representation of Subsite S1 – S5. Deep pockets of S1, S2 & S3 is shown here while S3 & S5 are located on the surface of the protein.
The substrate – binding pocket lies in the cleft between domain I and II [4]. It is comprised of Cys145 and His 41, called catalytic dyad [4] . There is no third catalytic element; it is compensated by a buried water molecule which forms H-bonds with the residue of His41 and the surrounding amino acids [7]. Another water molecule located within the active site of the enzyme established H-bonds with Phe140, His163, and Glu166 which stabilizes the oxyanion hole [7]. Besides the catalytic center (Cys145 & His41), there are three deeply buried subsites, S1, S2 & S4, with shallow subsites of S3 and S5 [3].
Why Mpro is an important drug target is because it is responsible for the maturation of itself and other important polyproteins. The Mpro (nsp5) encoded by the major ORF1ab, cleaves two overlapping polyproteins (pp1a & pp1ab) into 16 non-structural proteins which are important for viral replication and maturation [3].
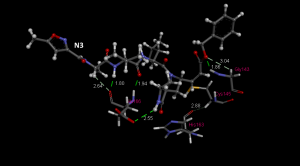
Figure 4. Hydrogen bonding interactions between inhibitor N3 with Gly143, Cys145, His163, Glu166 (PDB ID: 6LU7) indicating the importance of designing inhibitors that will interact with these residues. All distances are in angstrom (Å).
Hydrogen bonds between small molecule (inhibitor/ligand) is important to determine the specificity of the molecule/ligand to bind. The higher the binding affinity of the ligand, the higher the probability of the ligand will be as a potential inhibitor. Gly143 is the most attractive residue to form hydrogen bonds with ligands followed by Glu166, Cys145, and His163 [2]. This information indicates the importance of designing drugs that will interact with these residues.
The S1 Subsite.
The S1 subsite consists of His163, Glu166, Cys145, Gly143, His172 and Phe140. Ser1 in each one protomer interacts with Phe140 and Glu166 with the other protomer to stabilize the S1 subsite, a structural feature that is essential for catalysis [4]. The back wall of subsite S1 is created by the side chains of Phe140 and His163 [3].
The S2 Subsite.
The S2 subsite consists of Cys145, His41, and Thr25 [3]. These residues mainly involved in hydrophobic and electrostatic interactions. Subsite S2 is more hydrophobic than subsite S1, because it binds the hydrophobic residues Leu or Phe in substrate position P2 [3]. The base of subsite S2 were made of Tyr54 [3] . Subsite S2 is flanked by the π systems of His41 and the main chains connecting Asp187, Arg188 and Gln189 [3].

Figure 5. Hydrophobicity of the Subsite S1 – S5. S1, S2 & S4 are more hydrophobic compared to S3 & S5.
The subsites S3–S5.
The shallow subsites S3 – S5 comprises of Met49, His41, Met165, Glu166 and Gln189. The subsite (S3 – S5) can tolerate different functionalities [1]. The subsite S3 is located between residues Glu166 and Gln189 and is more than 9Å apart [3] .
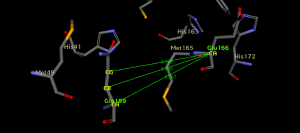
Figure 6. Respective distances in Å between Gln189 carbon atoms and one of Glu166 carbon atom (CG:Gln189 to CA:Glu166 = 9.168Å; CB:Gln189 to CA:Glu166 = 9.806Å; CA:Gln189 to CA:Glu166 = 9.567Å). The protein structure used is PDB ID: 6LU7.
Subsite S4 is a well defined binding pocket structure compared to subsites S3 and S5. Subsite S3 and S5 are located on the protein surface which made it more accessible to the bulk solvent (more hydrophilic). [3]
Subsite S5 is between Pro168 and residues Met165-His172 with Thr190- Ala194 acting as its base. The back wall of subsite S5 is formed by Leu167 and Phe185 creating a deep hydrophobic pocket of subsite S4. [3]
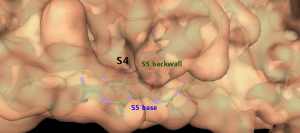
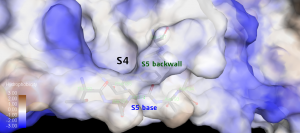
Figure 7. The Subsite S5 base residues and backwall creates a deep hydrophobic pocket of Subsite S4 .
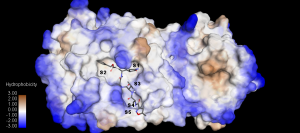
Figure 8. The inhibitor N3 accommodating the binding subsite S1-S5 (PDB ID:6LU7). Again S1, S2, & S4 are deep hydrophobic pockets while S3 & S5 is on the surface of the protein. The inhibitor N3 P groups occupies the respective subsites.
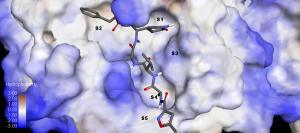
Figure 9. A closer view of the inhibitor N3 in the binding cleft according to the hydrophobicity of the binding pocket S1 – S5.
Conclusion.
- S1, S2 & S4 are deep hydrophobic pockets. To accommodate these pockets, inhibitors should be designed to have hydrophobic active groups.
- Residues Cys145, His41, and Thr25 mainly involved in hydrophobic and electrostatic interactions.
- S3 & S5 are shallow subsite located on the surface of the protein (hydrophilic). Inhibitors with corresponding P3 & P5 should match the criteria as hydrophilic groups.
- Gly143, Glu166, Cys145, & His163 are important residues for hydrogen bonding interactions with ligands/inhibitors. Inhibitors can be designed to have active groups interacting with these residues.
References:
[1] Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach, Salman Ali Khan, Komal Zia, Sajda Ashraf, Reaz Uddin & Zaheer Ul-Haq. Journal of Biomolecular Structure and Dynamics, Volume 39, 2021 – Issue 7.
[2] Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease, Hylemariam Mihiretie Mengist, Tebelay Dilnessa & Tengchuan Jin, Frontiers in Chemistry, 12 March 2021.
[3] Unusual zwitterionic catalytic site of SARS–CoV-2 main protease revealed by neutron crystallography,
Daniel W. Kneller, Gwyndalyn Phillips, Kevin L. Weiss, Hugh M. O’Neill, Leighton Coates, Andrey Kovalevsky, Journal of Biological Chemistry, VOLUME 295, ISSUE 50, P17365-17373, DECEMBER 2020.
[4] Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur, Zhenming Jin, Yao Zhao, Yuan Sun, Bing Zhang, Haofeng Wang, Yan Wu, Yan Zhu, Chen Zhu, Tianyu Hu, Xiaoyu Du, Yinkai Duan, Jing Yu, Xiaobao Yang, Xiuna Yang, Kailin Yang, Xiang Liu, Luke W. Guddat, Gengfu Xiao, Leike Zhang, Haitao Yang & Zihe Rao, Nature Structural & Molecular Biology, 27, pages529–532 (2020).
[5] Design of Wide-Spectrum Inhibitors Targeting Coronavirus Main Proteases, Haitao Yang, Weiqing Xie, Xiaoyu Xue, Kailin Yang, Jing Ma, Wenxue Liang, Qi Zhao, Zhe Zhou, Duanqing Pei, John Ziebuhr, Rolf Hilgenfeld, Kwok Yung Yuen, Luet Wong, Guangxia Gao, Saijuan Chen, Zhu Chen, Dawei Ma,corresponding, Mark Bartlam, and Zihe Rao, PLoS Biol. 2005 Oct; 3(10): e324.
[6] Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease, Wenhao Dai, Bing Zhang, Haixia Su, Jian Li, Yao Zhao, Xiong Xie, Zhenming Jin, Fengjiang Liu, Chunpu Li, You Li, Fang Bai, Haofeng Wang, Xi Cheng, Xiaobo Cen, Shulei Hu, Xiuna Yang, Jiang Wang, Xiang Liu, Gengfu Xiao, Hualiang Jiang, Zihe Rao, Lei-Ke Zhang, Yechun Xu, Haitao Yang, and Hong Liu, Science. 2020 Apr 22 : eabb4489.
[7] SARS-CoV-2 Mpro: A Potential Target for Peptidomimetics and Small-Molecule Inhibitors, Andrea Citarella, Angela Scala, Anna Piperno and Nicola Micale, Biomolecules 2021, 11(4), 607.
Notes:
1)all images are created using BIOVIA Discovery Studio 2021 Visualizer.
2)The PDB: 6LU7 used in this study/article was downloaded from the Protein Databank website.
3)*P: substrate/inhibitor residues; *S: protease binding sites. A detailed explanation of the terms P and S substrate-protease group can be found in this article.